Predicting surfactant phase behavior with a molecularly informed field theory
A novel multiscale simulation approach for investigating surfactants and self-assembly in complex systems
PIs and Institution
Kevin Shen, M. Scott Shell, Glenn H. Fredrickson, UC Santa Barbara
Achievement
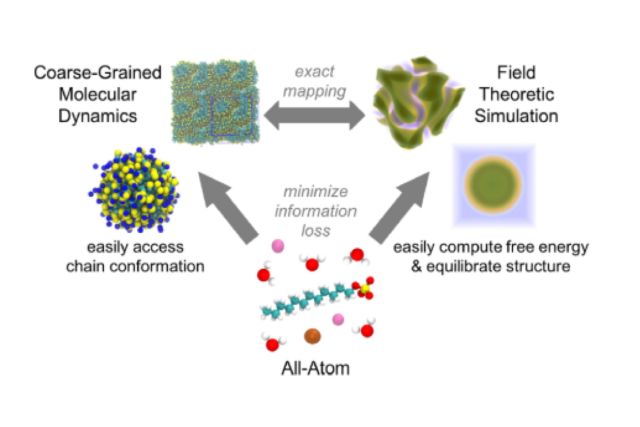
The study introduces a novel multiscale simulation approach for investigating surfactants and self-assembly in complex systems. Overcoming challenges in conventional molecular dynamics, it transfers chemical details from all-atom molecular dynamics, coarse-grained molecular dynamics, and field-theoretic simulations, which are respectively well-suited for describing chemical detail, simulating small-scale assemblies, and equilibrating large self-assembled structures. The method was successfully applied to sodium dodecylsulfate, reproducing both all-atom simulations and experimental data, allowing for the study of complex self-assembled structures like double or alternating gyroids, and reproduce salt effects on properties like aggregation number and shape transitions.
Importance of the Achievement
This research provides a systematic and efficient method for studying the self-assembly of surfactants in complex multicomponent systems. Surfactant self-assembly plays a crucial role in various industrial applications, such as formulation design and the development of nanomaterials. Traditional simulation methods often struggle to capture the intricate behavior of surfactants due to long equilibration times and the need for extensive computational resources. The proposed multiscale approach bridges the gap between all-atom simulations, which provide detailed chemical information, and coarse-grained simulations, which are computationally efficient. It allows for the exploration of complex 3D phases and the prediction of surfactant properties with high accuracy, facilitating the design and optimization of formulations in various industries. This research contributes to our understanding of surfactant behavior and offers a powerful tool for the development of innovative materials.
Synergies with BioPACIFIC MIP
This project made use of BioPACIFIC MIP computational facilities and Kevin Shen received support as a BioPACIFIC MIP Postdoctoral Fellow for its execution. The results have been integrated into a simulation workflow testbed available at: https://biopacificmip.org/platform/computation. This testbed offers an extensive toolkit for simulation workflows, bridging the gap between all-atom and field theoretic models, and is accessible to the wider community.